
GLEAM: Gene-set and cell-state exploration across space and time in R
![]()
![]()
![]()
GLEAM provides pathway/signature scoring, cell-state exploration, differential analysis after scoring, trajectory-aware mapping, and spatial transcriptomics analysis for both matrix-native and Seurat workflows. Built-in geneset examples focus on human and mouse, and custom genesets remain fully supported for other species.
Current scoring methods are standardized to canonical names: rank, mean, zscore, scaled_mean, robust_mean, ensemble, AddModuleScore, UCell, AUCell, ssGSEA, GSVA, singscore.
Installation
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
devtools::install_github("JamesWu7/GLEAM")Navigation: Documentation | Reference | Tutorials | Citation
Workflow highlights
Figures below are generated from scripts/GLEAM_homepage_showcase.Rmd via scripts/generate_homepage_figures.R.
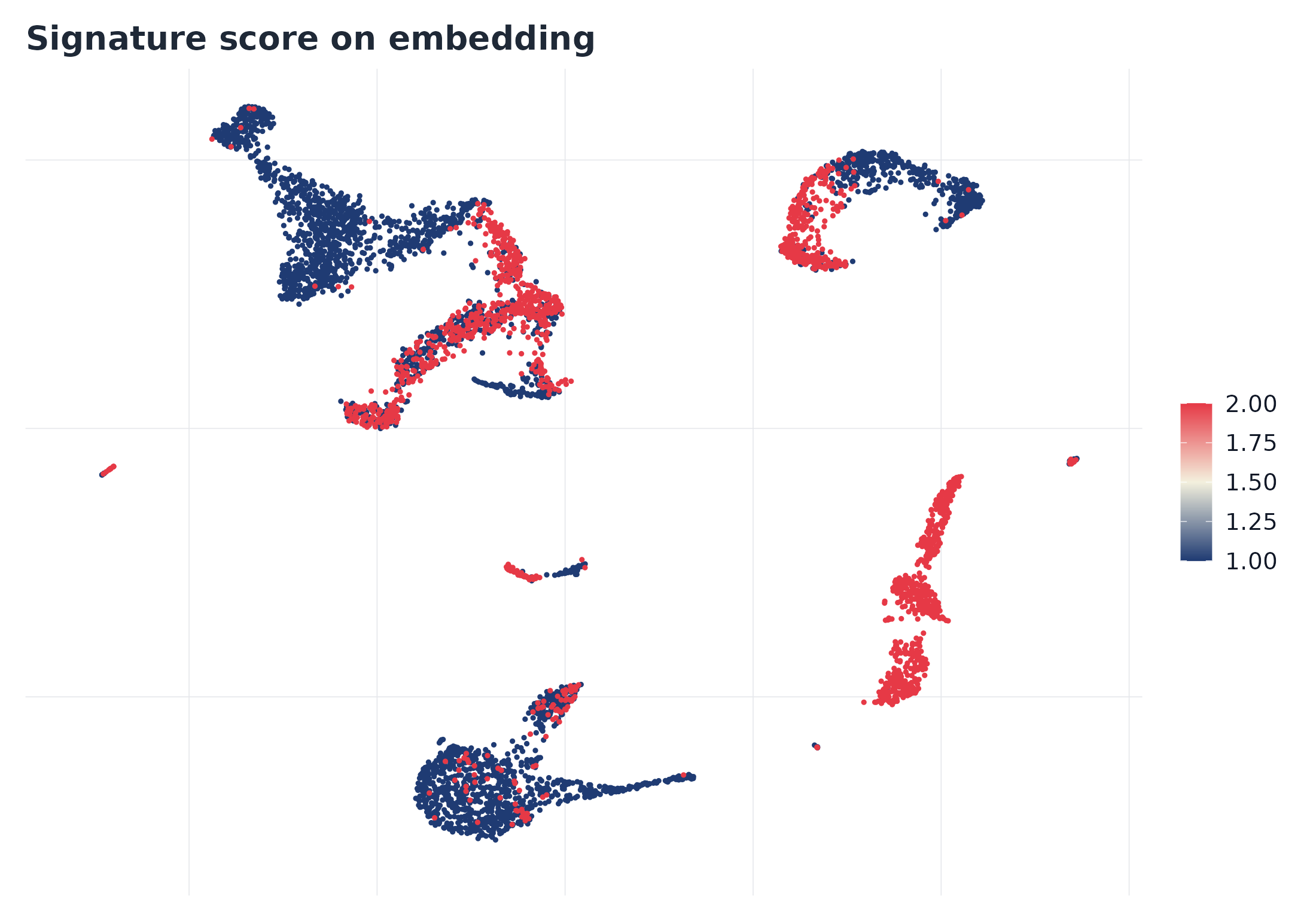
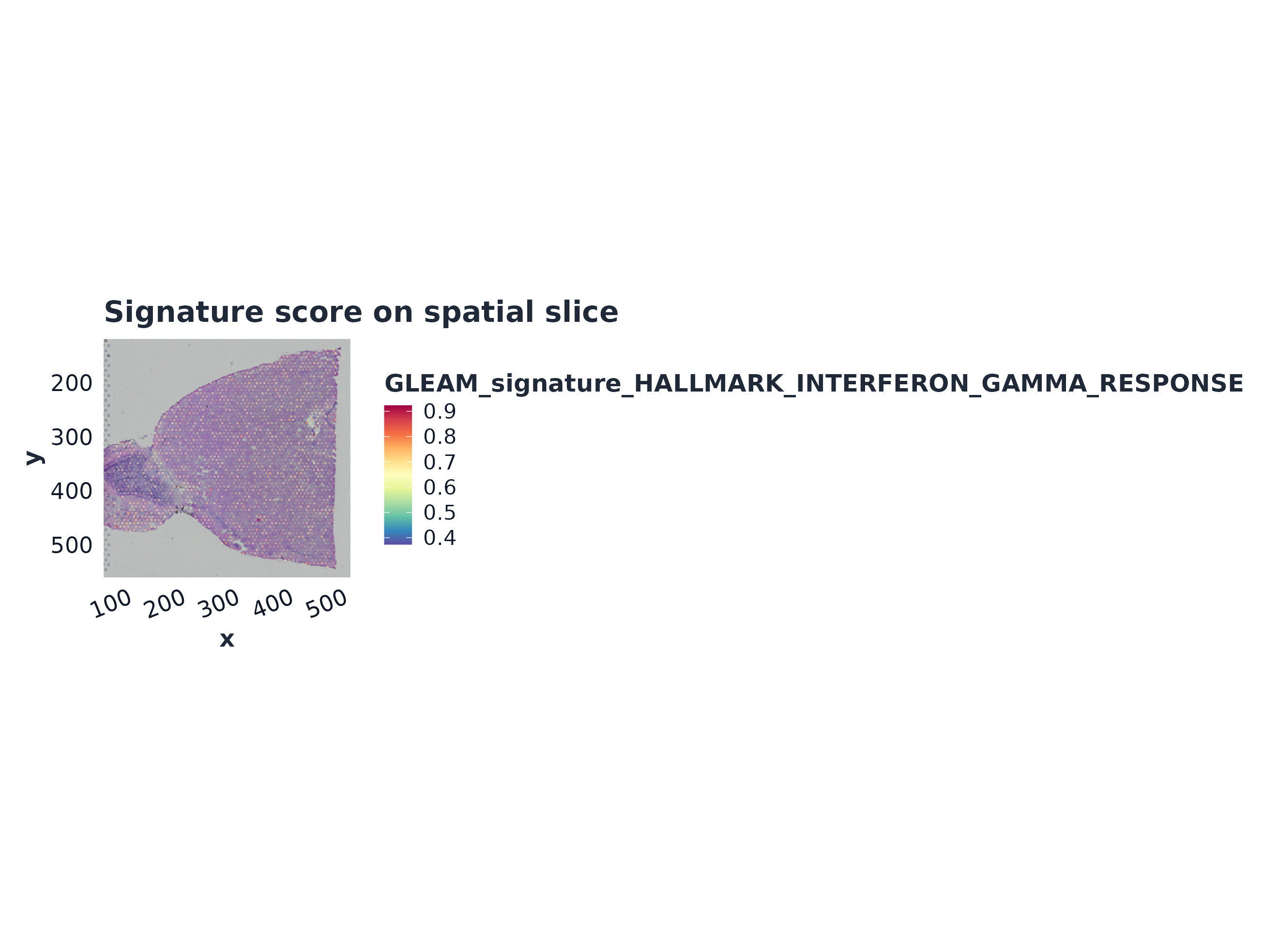
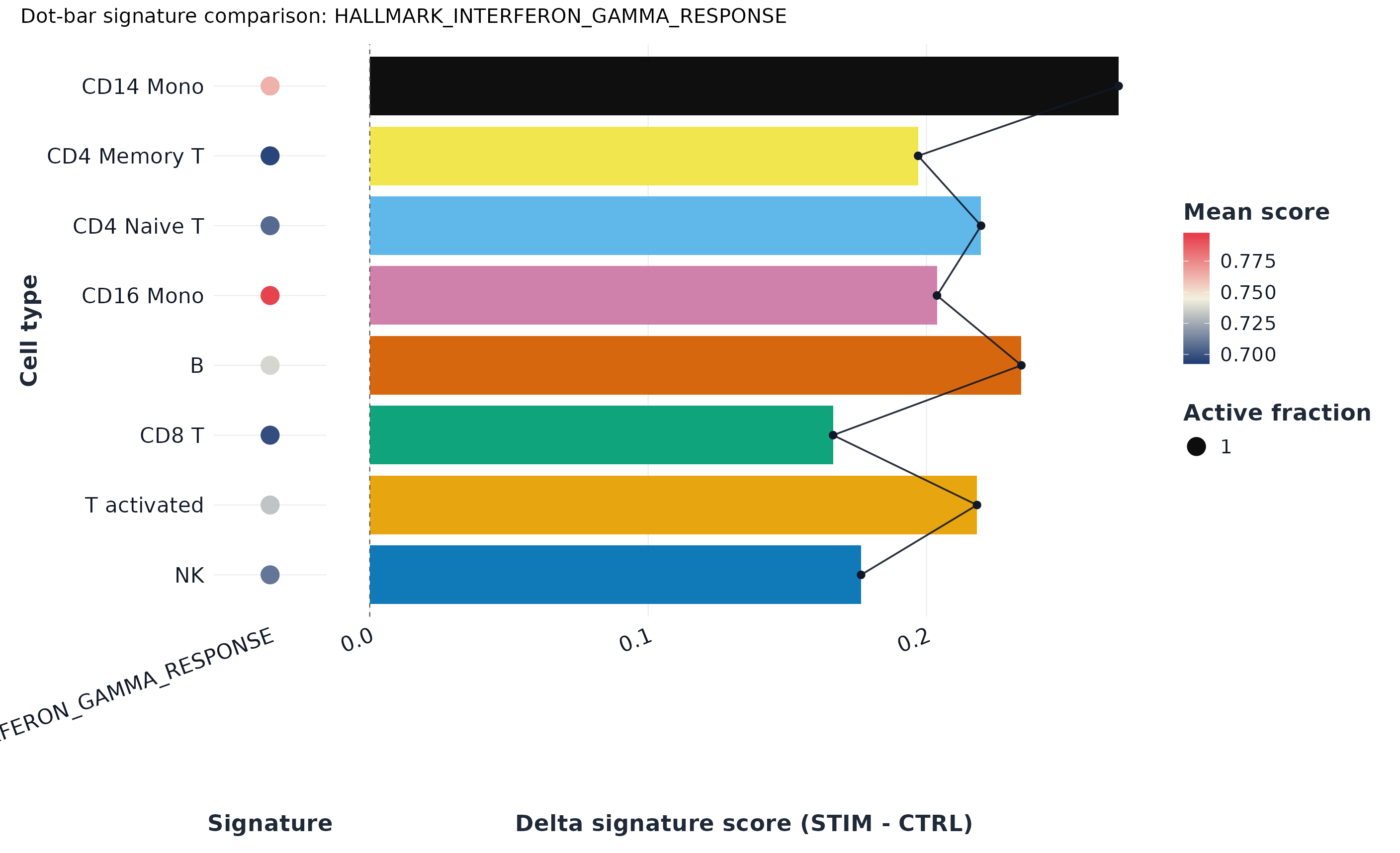
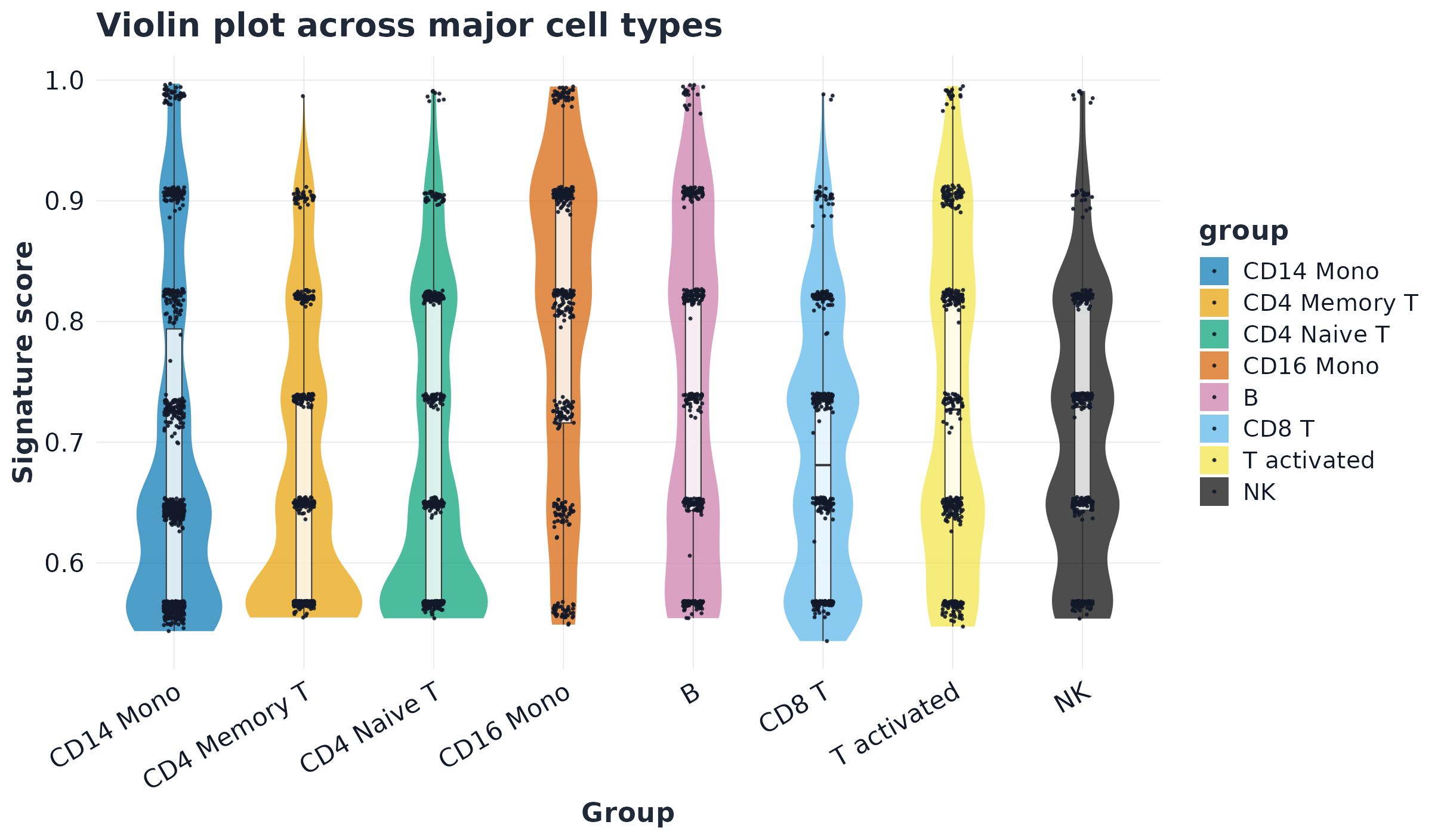
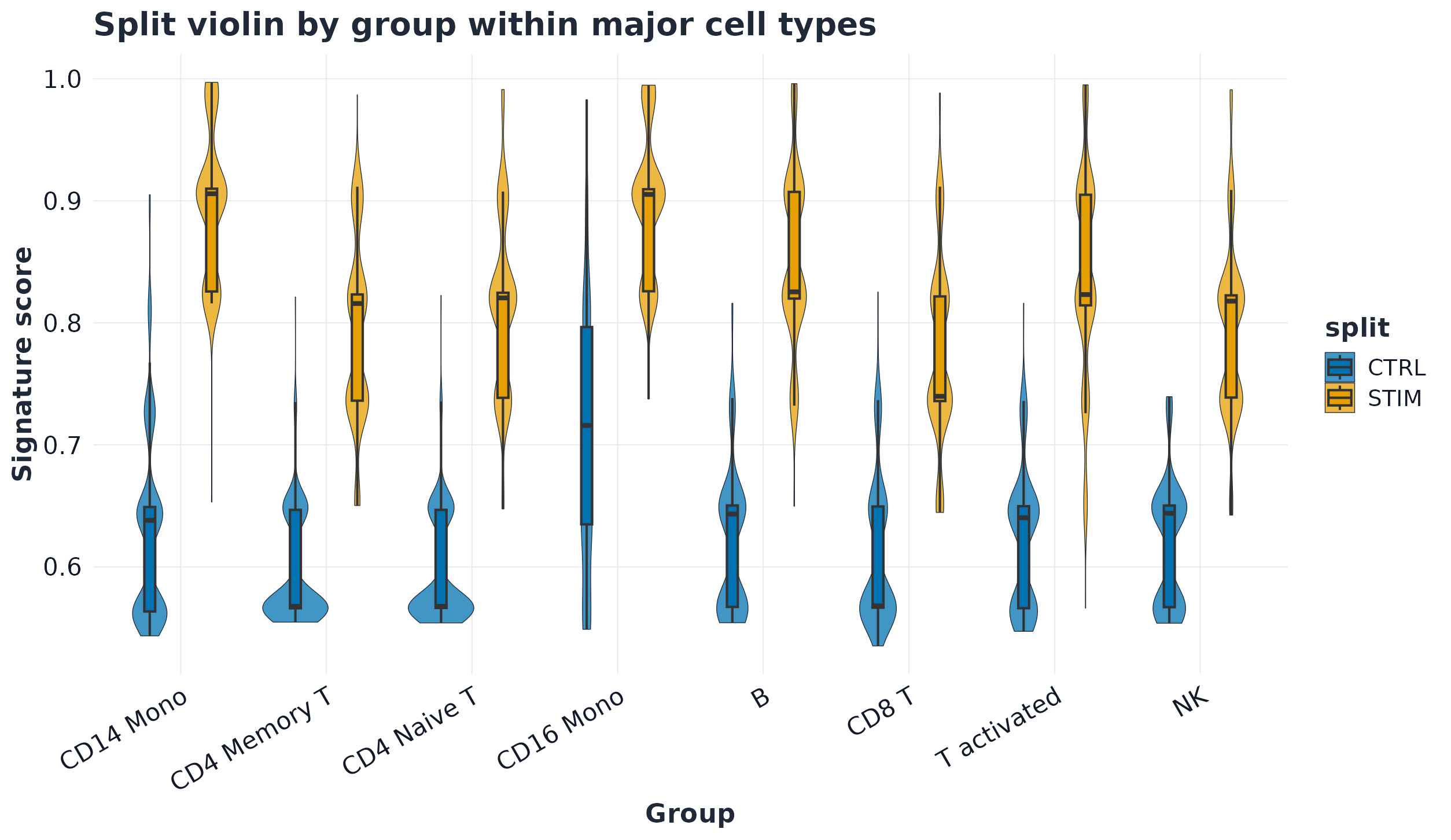
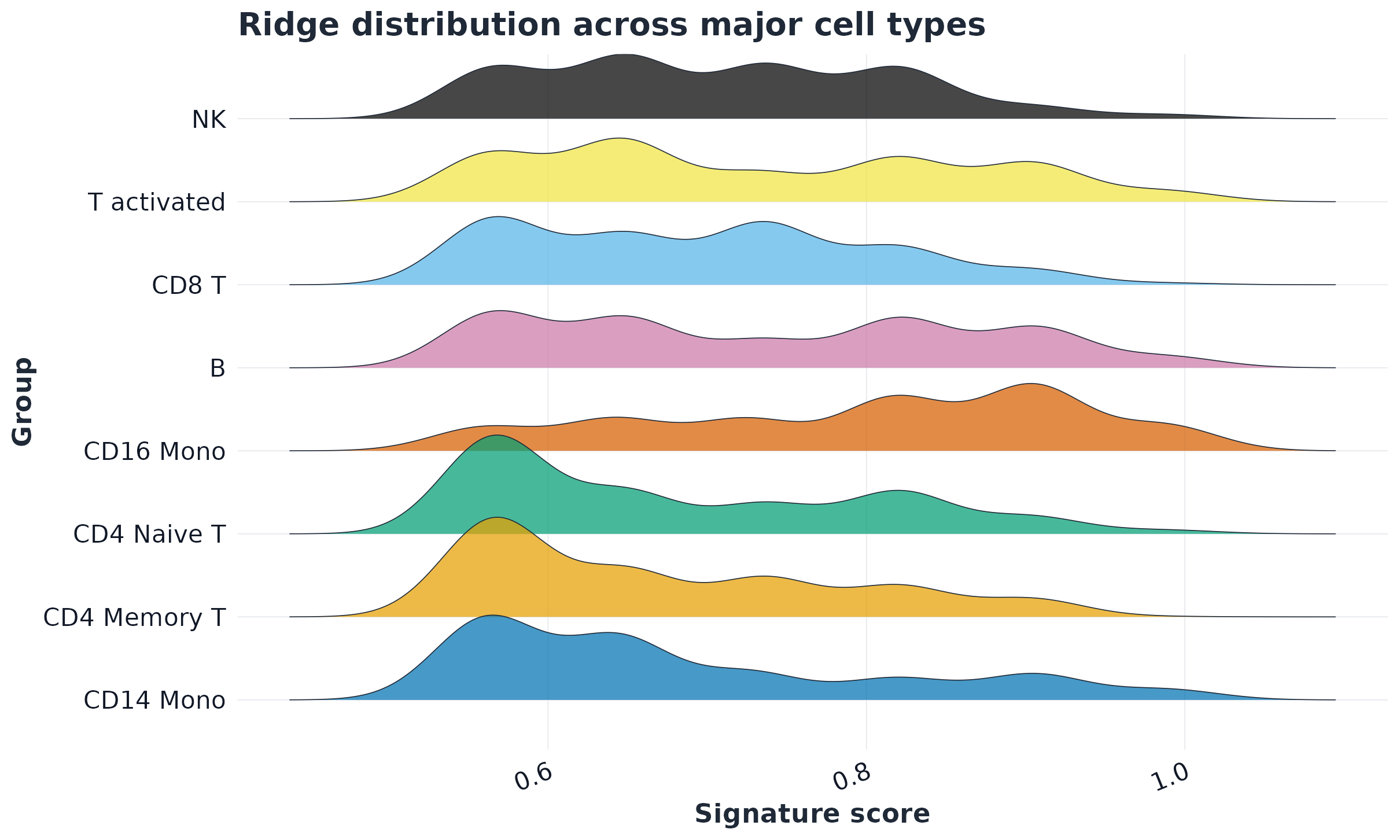
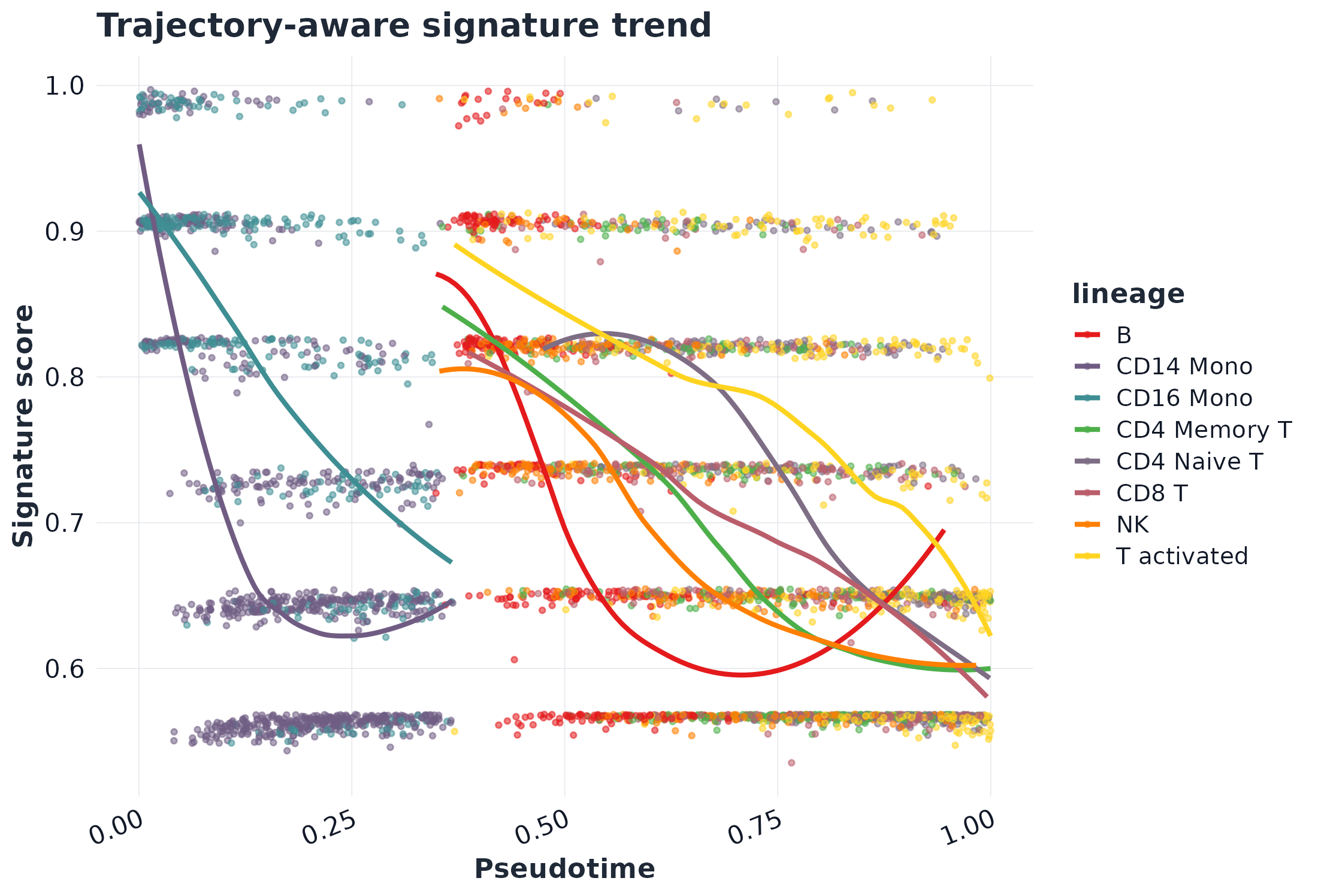
Quick start (Seurat scRNA-seq)
library(GLEAM)
library(Seurat)
ifnb_path <- system.file("extdata", "full_examples", "ifnb_seurat.rds", package = "GLEAM")
if (ifnb_path == "") ifnb_path <- file.path("inst", "extdata", "full_examples", "ifnb_seurat.rds")
seu <- readRDS(ifnb_path)
if (!"pca" %in% names(seu@reductions)) seu <- RunPCA(seu, verbose = FALSE)
if (!"umap" %in% names(seu@reductions)) seu <- RunUMAP(seu, dims = 1:20, verbose = FALSE)
meta_cols <- colnames(seu@meta.data)
group_col <- if ("stim" %in% meta_cols) "stim" else if ("group" %in% meta_cols) "group" else "orig.ident"
celltype_col <- if ("seurat_annotations" %in% meta_cols) "seurat_annotations" else if ("celltype" %in% meta_cols) "celltype" else "seurat_clusters"
seu$sample <- if ("orig.ident" %in% meta_cols) as.character(seu$orig.ident) else "sample_1"
hallmark_gs <- tryCatch(get_geneset("hallmark", source = "builtin", species = "human"), error = function(e) NULL)
if (is.null(hallmark_gs)) hallmark_gs <- tryCatch(get_geneset("hallmark", source = "builtin", species = "mouse"), error = function(e) NULL)
if (is.null(hallmark_gs)) hallmark_gs <- list(Signature_A = rownames(seu)[seq_len(min(30, nrow(seu)))])
sc <- score_signature(
object = seu,
geneset = hallmark_gs,
geneset_source = "list",
seurat = TRUE,
method = "ensemble",
min_genes = 3
)
res <- test_signature(sc, group = group_col, sample = "sample", celltype = celltype_col, level = "pseudobulk")
top_pw <- res$table$pathway[order(res$table$p_adj)][1]
if (is.na(top_pw) || !nzchar(top_pw)) top_pw <- rownames(sc$score)[1]
cell_levels <- names(sort(table(as.character(sc$meta[[celltype_col]])), decreasing = TRUE))
pal_cell <- setNames(get_palette("gleam_discrete", n = length(cell_levels), continuous = FALSE), cell_levels)
p1 <- plot_embedding_score(sc, signature = top_pw, object = seu, reduction = "umap")
p2 <- plot_violin(sc, signature = top_pw, group = celltype_col, palette = pal_cell, point_size = 0)
if (requireNamespace("patchwork", quietly = TRUE)) {
p1 + p2 + patchwork::plot_layout(ncol = 2)
} else {
p1
p2
}
Quick start (Seurat spatial)
library(GLEAM)
library(Seurat)
st_path <- system.file("extdata", "full_examples", "stxBrain_anterior1_seurat.rds", package = "GLEAM")
if (st_path == "") st_path <- file.path("inst", "extdata", "full_examples", "stxBrain_anterior1_seurat.rds")
st <- readRDS(st_path)
md_st <- st@meta.data
region_col <- if ("seurat_annotations" %in% colnames(md_st)) "seurat_annotations" else if ("region" %in% colnames(md_st)) "region" else "seurat_clusters"
if (region_col == "seurat_clusters") md_st$seurat_clusters <- paste0("cluster_", md_st$seurat_clusters)
st@meta.data <- md_st
gs_st <- NULL
for (sp in c("mouse", "human")) {
gs_try <- try(get_geneset("hallmark", source = "builtin", species = sp), silent = TRUE)
if (inherits(gs_try, "try-error")) next
gs_try <- lapply(gs_try, function(g) intersect(unique(as.character(g)), rownames(st)))
gs_try <- gs_try[vapply(gs_try, length, integer(1)) >= 3L]
if (length(gs_try) > 0L) {
gs_st <- gs_try
break
}
}
if (is.null(gs_st) || length(gs_st) == 0L) {
genes <- rownames(st)
n_take <- min(30L, length(genes))
gs_st <- list(Spatial_signature = unique(genes[seq_len(n_take)]))
}
sp <- score_signature(
object = st,
geneset = gs_st,
geneset_source = "list",
seurat = TRUE,
layer = "counts",
slot = "counts",
method = "rank",
min_genes = 3
)
top_sig <- rownames(sp$score)[1]
p1 <- plot_spatial_score(sp, signature = top_sig, object = st)
p2 <- plot_dot(sp, by = region_col)
if (requireNamespace("patchwork", quietly = TRUE)) {
p1 + p2 + patchwork::plot_layout(widths = c(2, 1))
} else {
p1
p2
}
Supported gene-set sources
-
builtin: in-package Hallmark-like and immune collections (human/mouse focus). -
list: user-provided named list. -
gmt: GMT file input viaread_gmt(). -
data.frame: tabular input withpathway+genecolumns. -
msigdb,go,kegg,reactome,wikipathways,biocarta: optional curated sources (dependency-gated, no silent internet-only behavior).
Visualization parameter guide
- Grouping/faceting:
group,group.by,split.by,region,sample,celltype. - Embeddings:
reduction = "umap"|"pca"|"tsne"in embedding/trajectory plots. - Spatial display: prefer
object = <Seurat spatial object>for native slice rendering;coords+ optionalimageremains available for matrix-mode overlays. - Style controls through theme helpers:
base_size,title_size,axis_text_size,legend_text_size,font_family,font_face,title_color,text_color. - Palette controls: plot-level
paletteplusget_palette(),scale_gleam_color(),scale_gleam_fill().
Full workflow tutorials
- Full scRNA workflow: GLEAM_full_scrna_workflow
- Full spatial workflow: GLEAM_full_spatial_workflow
Citation
- GitHub repository: https://github.com/JamesWu7/GLEAM
- R-native citation:
citation("GLEAM")
Suggested text for manuscripts:
GLEAM: Gene-set and cell-state exploration across space and time in R. R package (v0.2.0). Available at: https://github.com/JamesWu7/GLEAM.
BibTeX: